

Las cardiomiopatías son enfermedades del músculo cardíaco (miocardio), caracterizadas por hallazgos anormales del tamaño de las cámaras, el grosor de las paredes, o hallazgos sugestivos de disfunción sistólica o diastólica. Las cardiomiopatías se clasifican como primarias o secundarias de acuerdo con las guías de la asociación americana del corazón. Las primarias consisten en trastornos únicos
Las cardiomiopatías son enfermedades del músculo cardíaco (miocardio), caracterizadas por hallazgos anormales del tamaño de las cámaras, el grosor de las paredes, o hallazgos sugestivos de disfunción sistólica o diastólica.
Las cardiomiopatías se clasifican como primarias o secundarias de acuerdo con las guías de la asociación americana del corazón. Las primarias consisten en trastornos únicos o predominantemente confinados al músculo cardíaco, que tienen causas genéticas, no genéticas o adquiridas. Las cardiomiopatías secundarias son trastornos que tienen daño miocárdico como resultado de una enfermedad sistémica, anatómica o multi-orgánica.
Definición y Preámbulo:
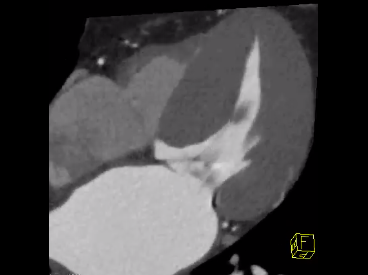
La miocardiopatía o cardiomiopatía hipertrófica (CMH) se define por la presencia de un aumento del grosor de las paredes del ventrículo izquierdo (con o sin hipertrofia del ventrículo derecho) que no se explica únicamente por condiciones anormales en la post-carga hemodinámica.
A su vez, CMH es un trastorno miocárdico con características histológicas que incluyen la hipertrofia de miocitos, desorganización de las miofibrillas, y fibrosis intersticial. Como otras cardiomiopatías, CMH puede presentarse como una entidad primaria o como parte de enfermedades sistémicas.
Anatómicamente, el grado de hipertrofia asimétrica o estenosis subaórtica puede causar obstrucción del flujo de la sangre en su salida del ventrículo izquierdo, generando la característica de poder ser obstructiva o no-obstructiva. En la variante obstructiva, otro factor que contribuye a este fenómeno es la creación de una zona de baja presión como consecuencia de un acelerado flujo sanguíneo a través de una región más angosta entre el tabique interventricular y las valvas mitrales (causando un efecto Bernoulli) que desencadena el movimiento anterior sistólico de la valva anterior perteneciente a la válvula mitral. A consecuencia de su patología, clínicamente se pueden apreciar arritmias, síncope, y muerte súbita.
Epidemiología:
La incidencia de CMH se estima entre 0.3 – 0.5 en 100,000 niños, que representan alrededor del 42% de los casos de la cardiomiopatía pediátrica primaria. La CMH es un poco más frecuente en el género masculino, y es más prevalente en niños de raza negra que en niños caucásicos o hispanos, y ocurre diez veces más frecuente en pacientes menores de 1 año en comparación con los niños mayores a esa edad. En adultos, de manera similar a la cardiomiopatía dilatada, CMH es mucho más frecuente y se estima a una prevalencia de al menos 1 entre 500 personas.
Causas genéticas y sistémicas de la CMH:
En el rango de edad pediátrica, las causas son variables y están relacionadas con la edad de su presentación. La CMH familiar es genéticamente heterogénea, y muchos de los genes involucrados se superponen entre los varios tipos de cardiomiopatías primarias. Investigaciones recientes sugieren que en la CMH familiar (separada de las enfermedades sistémicas y trastornos metabólicos) es común encontrar mutaciones en los genes que codifican proteínas del sarcómero y el citoesqueleto. Cada una de estas proteínas está codificada por uno o varios genes que proporcionan patrones específicos y fisiológicamente regulados al momento de expresión fenotípica.
-Las mutaciones más comunes en el sarcómero que ocasionan CMH incluyen las que codifican para las funciones de la cadena pesada de miosina, actina, tropomiosina, troponinas T, C e I, miopaladina, titina, miozenina, anquirina, teletonina, nexilina, entre muchas otras.
-Las mutaciones más comunes en el citoesqueleto que ocasionan CMH ocurren en los genes que codifican para integrina, junctofilina, PDZ, LIM, vinculina, y metavinculina.
-Otras causas comunes de CMH asociadas con enfermedades sistémicas incluyen:
1) Enfermedades metabólicas que pueden ser transmitidas con patrón autosómico dominante o recesivo. Ejemplos incluyen la enfermedad de Anderson-Fabry, mutaciones en los genes PRKAG2, la enfermedad de Danon y la enfermedad de Pompe; esta última con mayor proporción en niños y adolescentes.
2) Miopatías mitocondriales: causadas por mutaciones en el ADN nuclear o mitocondrial que se transmiten como rasgos autosómicos dominantes, autosómicos recesivos, ligados al cromosoma X, y patrones hereditarios mitocondriales. Algunos ejemplos son representados por mutaciones en genes que codifican para los complejos de la cadena respiratoria.
3) Enfermedad neuromuscular como la ataxia de Friedreich, y la miopatía por anormalidades en la nemalina y la desmina.
4) Síndromes de malformación, los más comunes son los causados por mutaciones en genes que codifican proteínas de la vía de la proteína quinasa activada por Ras/mitogen (MAPK) incluyendo síndrome de Noonan, síndrome LEOPARD (maculas cutáneas, Lentigines, anomalías de ECG, hipertelorismo ocular, estenosis Pulmonar, Genitales anormales, Retraso del crecimiento y sordera neurosensorial) y síndrome de Costello. La mayoría se diagnostican en la infancia, pero algunas formas más leves (particularmente el síndrome de Noonan) escapan a la detección temprana y se identifican más adelante en la vida.
5) Enfermedades infiltrantes, ejemplos comunes incluyen la amiloidosis cardíaca que produce un aumento progresivo en el grosor del miocardio ventricular izquierdo y derecho, el tabique interauricular y las válvulas. Las amiloidosis relacionadas con la cadena ligera (AL) y la transtiretina hereditaria (TTR) pueden afectar al corazón de forma aislada o con afectación multiorgánica, mientras que la amiloidosis TTR de tipo salvaje (senil) afecta predominantemente al corazón y al ligamento del túnel del carpo.
6) Trastornos endocrinos como la hipertrofia ventricular transitoria se ve en neonatos de madres con diabetes, incluso después de un buen control diabético durante el embarazo. En adultos, se ha descrito hipertrofia ventricular izquierda en asociación con feocromocitoma y acromegalia.
Características clínicas y diagnóstico:
El sello cardinal de esta enfermedad es la presencia de la hipertrofia de las paredes del ventrículo izquierdo, la hipercontractilidad miocárdica con un tamaño ventricular reducido, y la disfunción diastólica. El fenotipo a su vez también incluye fibrosis miocárdica, anomalías morfológicas del aparato de la válvula mitral, función coronaria anormal y anomalías electrocardiográficas. Debido a la diversa etiología de la enfermedad, la detección del aumento del espesor de la pared del ventrículo izquierdo que es inexplicable por las condiciones de post-carga debe iniciar una búsqueda sistemática de posibles causas subyacentes. En muchos pacientes, esta labor debe incluir pruebas de laboratorio, electrocardiografía, y en algunas circunstancias, análisis genéticos.
La construcción de un pedigrí familiar de tres a cuatro generaciones ayuda a confirmar un origen genético de la enfermedad e identifica a otros miembros de la familia que están en riesgo de desarrollo de la enfermedad. Las características específicas para tener en cuenta en los antecedentes familiares incluyen muertes cardíacas súbitas, insuficiencia cardíaca inexplicable, trasplante cardíaco, marcapasos/desfibriladores, y evidencia de enfermedad sistémica. El análisis de pedigrí también puede determinar el modo probable de herencia. La mayoría de las formas genéticas de CMH son de dominancia autosómica y, por lo tanto, se caracterizan por la presencia de individuos afectados en cada generación, con transmisión de padres de ambos sexos y un riesgo del 50% para la descendencia. La herencia ligada al cromosoma X debe sospecharse si los varones son los únicos o los más gravemente afectados y no hay transmisión de hombre a hombre. La herencia autosómica recesiva, el patrón menos común, es probable cuando ambos padres son portadores del gen afectado por lo que se puede ver asociada a consanguinidad. Cuando las mujeres, pero no los hombres, transmiten la enfermedad a niños de ambos sexos, se deben considerar mutaciones mitocondriales del ADN.
En un adulto, el diagnóstico se define cuando el grosor de la pared es igual o mayor a 15 mm en uno o más segmentos miocárdicos del ventrículo izquierdo, medidos por cualquier técnica de imagen (ecocardiografía, resonancia magnética cardíaca o tomografía computarizada), que no se explica únicamente por la post-carga. En niños, el criterio aún es falto de mayor definición, pero se contempla el diagnóstico cuando un paciente se encuentra con un espesor de pared ventricular izquierda más de dos desviaciones estándar superiores a la media de la población. Importante es tener en cuenta desafíos clínicos como los atletas en entrenamiento, la coexistencia de hipertensión arterial sistémica, y enfermedad valvular.
Tratamiento:
En ausencia de grandes ensayos aleatorizados, la terapia farmacológica se administra sobre una base empírica para mejorar la capacidad funcional, reducir los síntomas y prevenir la progresión de la enfermedad. En pacientes sintomáticos con obstrucción del tracto ventricular izquierdo, el objetivo es mejorar los síntomas mediante el uso de drogas, cirugía, o incluso ablación con alcohol. Por consenso, pacientes con obstrucción sintomática deben ser tratados inicialmente con beta bloqueadores (no vasodilatadores); los bloqueadores de los canales de calcio (no dihidropiridinas) deben ser usados con mucha precaución. Pacientes con obstrucción deben evitar la deshidratación y deben ser fomentados para evitar el sobre peso. Los dilatadores arteriales y venosos, incluidos los nitratos y los inhibidores de la fosfodiesterasa tipo 5, pueden exacerbar la obstrucción y deben evitarse si es posible.
La terapia en pacientes sintomáticos sin obstrucción se centra en el manejo de arritmias, la disfunción diastólica del ventrículo izquierdo y el tratamiento de angor pectoris. El uso de desfibriladores implantables debe ser considerado como prevención primaria o secundaria de acuerdo con las guías actuales en el manejo de CMH. Los pacientes con disfunción sistólica o diastólica progresiva y refractaria a la terapia médica pueden ser candidatos para un trasplante cardíaco. Actualmente, protocolos de investigación están enfocados en terapias con moduladores de la miosina como mavacampten; y terapias genéticas para modificar en DNA humano.
Referencias:
Kaski JP, Syrris P, Esteban MT, et al: Prevalence of sarcomere protein gene mutations in preadolescent children with hypertrophic cardiomyopathy. Circ Cardiovasc Genet 2:436–441, 2009.
Maron BJ, Peterson EE, Maron MS, et al: Prevalence of hypertrophic cardiomyopathy in an outpatient population referred for echocardiographic study. Am J Cardiol 73:577–580, 1994.
Morita H, Rehm HL, Menesses A, et al: Shared genetic causes of cardiac hypertrophy in children and adults. N Engl J Med 358:1899–1908, 2008.
Seidman JG, Seidman C: The genetic basis for cardiomyopathy: from mutation identification to mechanistic paradigms. Cell 104:557–567, 2001.
Blair E, Redwood C, de Jesus Oliveira M, et al: Mutations of the light meromyosin domain of the beta-myosin heavy chain rod in hypertrophic cardiomyopathy. Circ Res 90:263–269, 2002.
Carniel E, Taylor MR, Sinagra G, et al: Alpha-myosin heavy chain: a sarcomeric gene associated with dilated and hypertrophic phenotypes of cardiomyopathy. Circulation 112:54–59, 2005.
Kabaeva ZT, Perrot A, Wolter B, et al: Systematic analysis of the regulatory and essential myosin light chain genes: genetic variants and mutations in hypertrophic cardiomyopathy. Eur J Hum Genet 10:741–748, 2002.
Jay A, Chikarmane R, Poulik J, et al: Infantile hypertrophic cardiomyopathy associated with a novel MYL3 mutation. Cardiology 124:248–251, 2013.
He Y, Zhang Z, Hong D, et al: Myocardial fibrosis in desmin-related hypertrophic cardiomyopathy. J Cardiovasc Magn Reson 12:68, 2010.
Gudkova A, Kostareva A, Sjoberg G, et al: Diagnostic challenge in desmin cardiomyopathy with transformation of clinical phenotypes. Pediatr Cardiol 34:467–470, 2013.
Perry Elliot et all. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC), European Heart Journal, Volume 35, Issue 39, 14 October 2014, Pages 2733–2779
Towbin JA, Lipshultz SE: Genetics of neonatal cardiomyopathy. Curr Opin Cardiol 14:250–262, 1999
Lin G, Nishimura RA, Gersh BJ, Phil D, Ommen SR, Ackerman MJ, Brady PA. Device complications and inappropriate implantable cardioverter defibrillator shocks in patients with hypertrophic cardiomyopathy, Heart , 2009, vol. 95 (pg. 709-714)
Bernard J. Gersh et al. 2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy.
Kramer CM et al. Hypertrophic Cardiomyopathy Registry: The rationale and design of an international, observational study of hypertrophic cardiomyopathy. Am Heart J. 2015 Aug;170(2):223-30. doi: 10.1016/j.ahj.2015.05.013. Epub 2015 May 22.
James D. Wilkinson et al. The Pediatric Cardiomyopathy Registry and Heart Failure: Key Results from the First 15 Years. Heart Fail Clin. PMC 2011 Oct 1.






































Deje un comentario
Registrese para comentar. Sus e-mail no será publicados