

– El extraordinario camino del conocimiento científico – La noción del corazón derecho excesivamente adiposo, como es el caso ahora bien aceptado de la que fue establecida como una nueva entidad clínica después de un extraordinario camino del conocimiento científico que comenzó en 1973 en el hospital de la Pitié-Salpêtrière en Paris con la cirugía
– El extraordinario camino del conocimiento científico –
La noción del corazón derecho excesivamente adiposo, como es el caso ahora bien aceptado de la que fue establecida como una nueva entidad clínica después de un extraordinario camino del conocimiento científico que comenzó en 1973 en el hospital de la Pitié-Salpêtrière en Paris con la cirugía de un hombre de 65 años sin enfermedad coronaria con episodios recurrentes de taquicardia ventricular que pareció originarse desde el ventrículo derecho (VD). El cirujano Gérard Guiraudon observó en el transoperatorio una dilatación moderada e hipocinesia del VD, cubierto por una prominente capa de grasa. El mapeo epicárdico por Guy H. Fontaine y Robert Frank, registró en el sitio potenciales tardíos alejados del QRS de superficie revelando que el “sitio de origen” de la taquicardia estuvo en la pared libre del VD. La ventriculotomía simple observó histológicamente una transformación adiposa de la pared con persistencia de una capa fina de endocardio.
El equipo se convenció que estaban ante una nueva patología del VD con características prominentes en pacientes con un perfil consistente, el caso fue uno de cuatro publicados en el libro The Conduction System of the Heart del profesor Hein J. Wellens en 1976.1 Se atribuye a Guy H. Fontaine el mérito de haber adoptado el termino “Displasia Arritmogénica del Ventrículo Derecho” publicado en el libro Reentrant Arrhythmias: Mechanisms and Treatment del profesor Henri Kulbertus en 1977.2 Este padecimiento fue incluido en la nueva clasificación de las cardiomiopatías de la OMS3 en 1996 y se nombró “Cardiomiopatía Arritmogénica del Ventrículo Derecho/Displasia” (CAVD/D) en 2010.4
La CAVD/D es probablemente, dentro del campo de la cardiología, la única enfermedad más compleja de diagnosticar. En esta publicación del Dr. Jorge Elias Neto, del Hospital Vitória Apart en Serra, Espírito Santo, Brasil y de los Drs. Joelci Tonet, Robert Frank y Guy Fontaine del Hospital Pitié-Salpêtrière, Paris, Francia con toda la experiencia de este grupo desde la década de 1970 se dieron a la tarea de revisar ampliamente los avances significativos en el conocimiento de la etiopatogenia con sus características histopatológicas, genéticas y moleculares, la presentación clínica e historia natural, el diagnóstico clínico con base en los criterios actuales del ECG y exámenes complementarios de imágenes, la estratificación del riesgo y las modalidades terapéuticas después de 40 años en el diagnóstico de esta entidad clínica.5
Con las primeras publicaciones electrocardiográficas y anatomo-clínicas,6, 7 y la experiencia acumulada de 1973 a 1980 del grupo de arritmias del hospital de la Salpêtrière y Jean Rostand en Francia, Frank I. Marcus, Guy H. Fontaine y cols. publican en 1982 la primera descripción sistemática sobre el diagnóstico y tratamiento de la CAVD/D.8
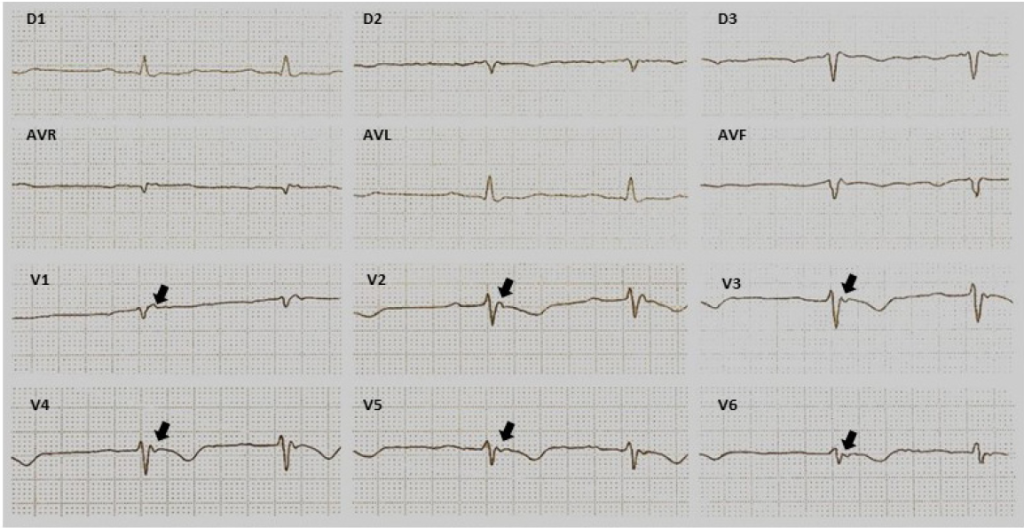
Desde entonces, mucho se ha aprendido en todos los aspectos de esta enfermedad. En los 1980 sobresalieron los reportes sobre las formas familiares de la enfermedad y variantes como la enfermedad de Naxos. En el periodo de 1990 surgieron las primeras series clínicas que llamaron la atención sobre el papel del esfuerzo físico como disparador de arritmias, siendo una causa importante de muerte súbita en los jóvenes, notablemente en los deportistas. Un avance significativo en 1994 fue la propuesta de criterios para facilitar el diagnóstico con base en el ECG, características estructurales e histológicas de la enfermedad, así como arritmias e historia familiar. Estos criterios fueron divididos en mayores o menores, de acuerdo a la especificidad para el diagnóstico de CAVD/D.9
Sin duda uno de los principales avances en el inicio del 2000 fue el descubrimiento de las mutaciones en el gen de Plakoglobina (locus 17q21) que codifica para los desmosomas, estructuras complejas de adhesión intercelular que se encuentran en los cardiomiocitos y las células epiteliales estratificadas de la piel y que resultó en las bases genéticas de la CAVD/D.10 Las alteraciones de los desmosomas relacionados con remodelación de las uniones gap son seguidas por desacoplamiento mecánico y eléctrico que terminan en muerte celular y reemplazo fibro-graso patognomónico. Actualmente, una mutación patogénica puede ser identificada en más del 60% de los sujetos afectados. Consistente con esto, es que las pruebas genéticas han surgido como una herramienta diagnóstica importante y juegan un papel crítico en la evaluación de familiares de primer grado. De manera general, los primeros criterios para el diagnóstico de CAVD/D se basaron principalmente de casos índice sintomáticos y etapas avanzadas de la enfermedad, estos criterios eran altamente específicos para el diagnóstico, pero de baja sensibilidad.9 La investigación clínica subsecuente en familias de CAVD/D y el descubrimiento de las mutaciones causales de la enfermedad aumentaron el conocimiento en la etiología y patogénesis de la enfermedad.
Por tanto, una nueva fuerza de tarea introdujo modificaciones a los criterios de 1994 implementando nuevos criterios.4 Estos criterios probaron ser altamente específicos subieron de menor a mayor y nuevos criterios sobre el retardo de activación y morfología de la taquicardia ventricular, así como criterios genéticos fueron agregados. Además, parámetros cuantitativos tanto de modalidades de imágenes como histopatológicos fueron introducidos y definidos con base en la comparación con sujetos sanos.4 Múltiples estudios en diferentes cohortes demostraron que los cambios ECG son los indicadores más tempranos y sensibles de CAVD/D y junto con los criterios genéticos han mejorado importantemente el diagnóstico. Hemos aprendido mucho sobre la importancia del ejercicio tanto en el desarrollo de CAVD/D en sujetos susceptibles y en el conocimiento del curso de la enfermedad, esto ha resultado en una mayor identificación de individuos afectados y contribuyen mejor a la prevención tanto de taquiarritmias ventriculares como muerte súbita. El desafío clínico en el paciente individual reside ahora principalmente en detectar las etapas tempranas, cuando no hay una expresión fenotípica manifiesta y la estratificación del riesgo. El esfuerzo debe dirigirse hacia aquellos que se beneficiaran más, al tiempo que se implementa una estrategia “de observación” en aquellos de bajo riesgo. El objetivo final del cardiólogo es ahora proporcionar un enfoque específico adaptado a cada paciente con CAVD/D.
- – Ver link AQUI






































4 Comments
Job Cortés O
septiembre 4, 2019, 12:13 amApreciado Dr. Enrique Velázquez Rodríguez, por este medio le saludo muy cordialmente. Leí con mucho entusiasmo la semblanza que usted hace del Gran médico Dr. Enrique Cabrera Cosío (http://someec.mx/pioneros/DR.-ENRIQUE-CABRERA-COSIO-Biografia.pdf). Agradecería mucho su ayuda para obtener más bibliográfica de la vida, PENSAMIENTO y obra del extraordinario hombre que fue el Dr. Enrique Cabrera Cosío. Gracias. Atte. Job Cortes O
REPLYVICENTE SANCHEZ CRESPO
abril 27, 2024, 6:16 pmExcelente screening de esta nueva nosologia,debo mencionar que en esa misma decada 70 .80 en nuestro pais Ecuador ciudad Guayaquil mi estimado profesor ,Dr,Luis Carvaja Huerta ,Jefe del servicio de Dermatologia del Hospital Luis Vernaza ,realizo la exaustiva investigacion de pacientes ,niños de seco masculino que presentaban otor fenotipo con penetrancia y varibilidad distinta con la triada clasica de pelo lanoso,hipérquearosis palmo plantar ,palpitaciones,con ECG trastonro de conducion av bloqueos brhh y t negativas anterolaterales ,provenientes de la prov.Los Rios ,EN GRUPO FAMILAR,con tet genetico positivo realizzdo en ese entonces EE.UU con positivo ,DSP´,posteriorment publicado en la Revista Dermatologia ,por lo que hoy recibe el nombre de un medico Guayaquileño con otro fenotipo y penetrancia variablñidad de expresion en su honor conocido como sindrome de Carvajal como una nueva nosologia cardiocutanea o desmosopatia, atte .CECP CENTER OF EXCELLENCE CARDIOGENETIC AND PEDIATRIC Dr.Vicente Sanchez Crespo Cardiologo Genetista UCAM España UCSG IESS
REPLYactualmente se esta tratando ademas del tratamiento cardiovascular ,con terapia genetica la desmosopatia com mARN 184 ,en ensayos efectividad y seguridad al igual que otras patologias con base geneticas representa nel 60 % alteranciones desmosomas uniona adherentes , sarcolema ,conexina 43 canales del na ,verbigraci :mevacamten para CMHO ,eternepleser disptrofia ,patisiran inotersen vutrisiran ,acoramidis para amiloidosis cardiaca nueva casgevy
VICENTE JAVIER SANCHEZ CRESPO
mayo 5, 2024, 12:06 pmEn nuestros país en las mismas décadas de los 70 u 80 el eminente y estimado Prof.Dr.Luis Carvajal Huerta realizaba la magistral descripción de un grupo familiar y en población infantil de esta patología con la triada de un algoritmo diseñado por Nosotros ALGORITMO SIND CARVAJAL.oeko latoso.hiperqueratosus pal.o plantar .cardiomipatia arritminogena…en esta población infantil y presentados con test genéticos en USA .lo que hoy lleva el nombre de un Médico Ecuatoriano sind Carvajal…..en el conocimiento actualizado de cardiogenetica y actualmente en ensayos con mARN 184…..remodelar el desacoplamiento de estas desmosopatia …merecido homenaje no postulo a nuestro querido profesor eminente jefe Dermatologia de ese entonces en el Hospital Luis Vernaza..Guayaquil Ecuador….atte.Dr.Vicente Sanchez Crespo Cardiologo EXPERTO en Genetica Médica y Genomic UCAM España. CECP CENTER OF EXCELLENCE CARDIOGENETC AND PEDIATRIC ……0994723819. 0991723505…..Atendemos pacientes de nuestro país y del exterior ASESORIA ACADEMICA INTERNACIONAL PRESTIGIOSAS UNIVERSIDADES DE EUROPA ASIA Y EE.UU. SALUDOS CORDIALES
REPLYVICENTE JAVIER SANCHEZ CRESPO
mayo 5, 2024, 12:06 pmEn nuestros país en las mismas décadas de los 70 u 80 el eminente y estimado Prof.Dr.Luis Carvajal Huerta realizaba la magistral descripción de un grupo familiar y en población infantil de esta patología con la triada de un algoritmo diseñado por Nosotros ALGORITMO SIND CARVAJAL.oeko latoso.hiperqueratosus pal.o plantar .cardiomipatia arritminogena…en esta población infantil y presentados con test genéticos en USA .lo que hoy lleva el nombre de un Médico Ecuatoriano sind Carvajal…..en el conocimiento actualizado de cardiogenetica y actualmente en ensayos con mARN 184…..remodelar el desacoplamiento de estas desmosopatia …merecido homenaje no postulo a nuestro querido profesor eminente jefe Dermatologia de ese entonces en el Hospital Luis Vernaza..Guayaquil Ecuador….atte.Dr.Vicente Sanchez Crespo Cardiologo EXPERTO en Genetica Médica y Genomic UCAM España. CECP CENTER OF EXCELLENCE CARDIOGENETC AND PEDIATRIC ……0994723819. 0991723505…..Atendemos pacientes de nuestro país y del exterior ASESORIA ACADEMICA INTERNACIONAL PRESTIGIOSAS UNIVERSIDADES DE EUROPA ASIA Y EE.UU. SALUDOS CORDIALES
REPLY