

Muchos errores existen en la práctica diaria de como medir u acerca de este tópico, aca una breve revision y ejemplos electrocardiograficos. Esperemos que les sea de utilidad y a la espera de comentarios como tambien de temas deseados. Dr. Ricardo Pignatelli. El síndrome de QT largo congénito (SQTL) es una enfermedad hereditaria caracterizada por una disfunción
Muchos errores existen en la práctica diaria de como medir u acerca de este tópico, aca una breve revision y ejemplos electrocardiograficos. Esperemos que les sea de utilidad y a la espera de comentarios como tambien de temas deseados. Dr. Ricardo Pignatelli.
El síndrome de QT largo congénito (SQTL) es una enfermedad hereditaria caracterizada por una disfunción de los canales iónicos cardíacos (canalopatía), que provoca un intervalo QT corregido (QTc) prolongado en el electrocardiograma (ECG) basal, asociado a una mayor predisposición de taquicardia ventricular polimórfica y muerte súbita en pacientes jóvenes sin cardiopatía estructural. 1
El SQTL tiene una prevalencia aproximada de 1:2000 niños nacidos vivos. Se identificaron mutaciones genéticas que causan SQTL en un 43% y un 29% de los pacientes menores de un año que presentaban valores de QTc mayores a 470 y 460 milisegundos (mseg) respectivamente. 2 Cabe aclarar que este dato no toma en cuenta aquellos pacientes con fenotipo negativo (valores normales de QTc) y genotipo positivo, con lo cual puede estimarse una prevalencia aún mayor. En el año 1995, se identificaron los primeros tres genes responsables del SQTL 3, estudios genéticos moleculares han revelado 13 variantes genéticas de SQTL causadas por mutaciones que codifican proteínas del canal de potasio, sodio, factores relacionados con el canal de calcio y proteínas adaptadoras de membrana. Los tipos más frecuentes son 3. Los pacientes con SQTL Tipo1, SQTL Tipo 2, y genotipos del SQTL Tipo 3 con mutaciones que involucran KCNQ1, KCNH2 y SCN5A constituyen más del 92% de los pacientes con SQTL genéticamente confirmado. Hasta un 20% -25% de los pacientes con LQTS siguen siendo genéticamente negativos. 4 El diagnóstico de LQTS se basa principalmente en la medición del intervalo QTc para la frecuencia cardíaca (QTc) usando la fórmula de Bazett. Se deben excluir causas secundarias de prolongación QTc que pueden ocurrir con ciertos fármacos, afecciones cardíacas adquiridas, desequilibrio electrolítico y dietas desequilibradas. Se ha establecido un sistema de puntuación (criterios de Schwartz) 5 de probabilidad diagnóstica de SQTL, que representan una importante guía en la evaluación inicial de los casos potenciales. Utilizan una puntuación del 1 al 9 según la historia familiar y los hallazgos clínicos y electrocardiográficos. Si el índice de puntuación es ≤ 1, la probabilidad de presentar la enfermedad es baja, si es 2-3 la probabilidad es intermedia, y si ≥ 4, es alta. 5,6(Tabla 1) 
Tabla 1. Escala de puntuación de Schwartz
El cuadro clínico de presentación es muy variable desde pacientes asintomáticos hasta síncope, convulsiones y muerte súbita. Se transmite al 50% de su descendencia con penetrancia variable. Previo a la clasificación acorde a los datos genéticos se caracterizaban en cuadros homocigotas con alto riesgo de muerte súbita que presentaban sordera congénita (Síndrome de Jerve-Lange Nielsen) y los heterocigotas de presentación clínica variable (Síndrome de Romano- Ward). Respecto al diagnóstico prenatal de síndrome de QT largo, la bradicardia fetal puede ser una de las primeras manifestaciones clínicas del SQTL y puede estar asociado a bloqueo AV o pseudo bloqueo AV (SQTL Tipo 2 y 3) que se esclarecerá en el ECG posnatal. En series retrospectivas se ha documentado que hasta un 70% de los pacientes diagnosticados en la infancia tiene este antecedente, que suele ir acompañado de hidrops fetalis. 7 La medición del intervalo QT representa la duración de la repolarización ventricular y se mide desde el inicio de la onda Q hasta el final de la onda T al regresar a la línea de base del ECG (Fig.1). Debe realizarse preferentemente en las derivaciones DII o V5, donde tiene mayor poder predictivo. 8,9 La fórmula de Bazett se utiliza para corregir la duración del intervalo de acuerdo con la frecuencia cardiaca (QTc = QT/√ RR, expresado en segundos).10 Se recomienda la medición manual y sistemática de este intervalo, en menos del 40% de los médicos no cardiólogos, menos del 50% de los cardiólogos y más del 80% de los arritmólogos supieron medirlo correctamente según un estudio multicéntrico.11 Es un intervalo dinámico y los límites normales dependen de varios factores. Si bien se ha considerado anormal un intervalo QTc ≥ 450 ms en los varones y ≥ 460 ms en las mujeres (≥ 470 ms ante la presencia de arritmia sinusal respiratoria), en este rango podemos encontrar tanto a portadores de mutaciones como a sujetos sanos.
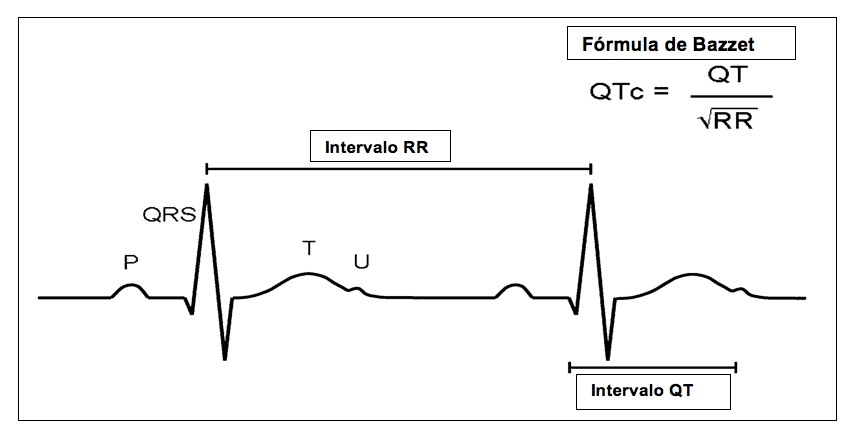
Fig. 1 Medición del intervalo QT en el ECG
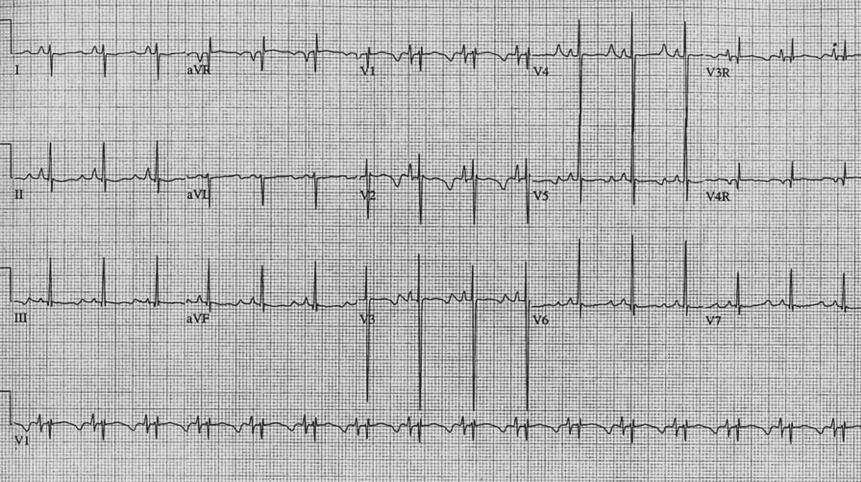
Los pacientes con SQTL pueden presentar múltiples alteraciones en la repolarización ventricular manifestándose en la onda T del ECG (Fig. 2), mostrando como máxima expresión de inestabilidad eléctrica la alternancia de la onda T (Onda T de polaridad variable latido a latido) Fig.3 . Los pacientes con SQTL pueden cursar con signos de disfunción del nódulo sinusal, bradicardia y/o pausas.12 Los subtipos SQTL1 y SQTL3, particularmente este último, presentan con frecuencia bradicardia sinusal. El bloqueo AV 2:1 o pseudo bloqueo AV 2:1 (por la prolongación exagerada del potencial de acción y período refractario ventricular, el siguiente impulso procedente de la actividad sinusal es bloqueado por encontrar a los ventrículos aún en período refractario) es una manifestación infrecuente pero de mal pronóstico, que puede presentarse desde la etapa fetal en forma de bradicardia persistente (Fig.4). La incidencia ha sido comunicada en el 4-5% y se asocia con una alta mortalidad a pesar del tratamiento con bloqueadores beta y/o marcapasos. 12,13
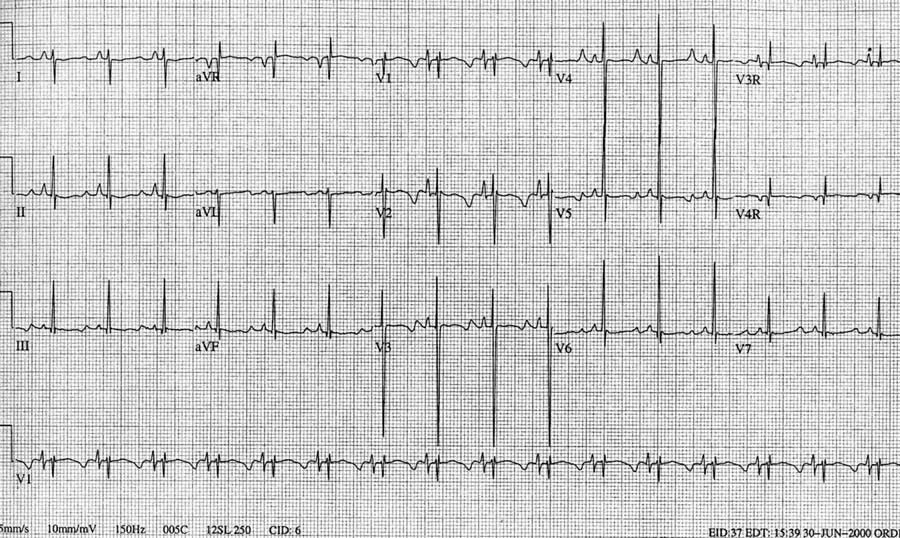
Fig. 2 Síndrome de QT largo

Fig. 3 Síndrome de QT largo y alternancia de la onda T.

Fig 4. QT Largo con pseudo bloqueo AV 2:1
La arritmia ventricular característica del SQTL es la conocida torsade de pointes, es una taquicardia ventricular polimórfica por reentrada, caracterizada electrocardiográficamente por un giro continuo del eje del QRS. Se presenta cuando el intervalo QT se prolonga, puede originar fibrilación ventricular y muerte súbita. Si esto no sucede, el paciente puede experimentar síncope o incluso, si el episodio es breve, puede pasar desapercibido. Según los subtipos en SQTL Tipo 1 los eventos son gatillados por estrés físico, natación y son más frecuentes en varones, en el SQTL Tipo 2 por ruidos repentinos o sustos durante la noche y es más frecuente en mujeres adultas con mayor vulnerabilidad durante el período post parto y en el SQTL Tipo 3 durante el sueño, más frecuente en varones. Debido a esto se investiga el rol del SQTL (sobre todo tipo 3 y 2) en el síndrome de muerte súbita del lactante. En el seguimiento es importante realizar Holter y ergometría para evaluación pronóstica y de la terapia antiarrítmica. Los pacientes con SQTL no suelen alcanzar la frecuencia máxima esperada calculada para la edad. Asimismo, el intervalo QT al esfuerzo puede tener un comportamiento paradójico, alargándose en lugar de acortarse. Los pacientes con SQTL Tipo1, además de no llegar a la frecuencia cardiaca máxima calculada para la edad, con frecuencia alargan el intervalo QTc, los SQTL Tipo 2 suelen alcanzar la frecuencia cardiaca esperada y prolongar sólo discretamente el intervalo QTc, o incluso no prolongarlo. Los pacientes con SQTL Tipo3 tienen, en general, una respuesta de acortamiento normal del intervalo QTc.14 La evaluación genética en el SQTL es de utilidad en el tratamiento de los enfermos, en particular en los casos de alto riesgo y su aplicación es importante para consejo genético. Respecto al tratamiento los betabloqueantes son la terapia de primera línea y están indicados en aquellos pacientes con SQTL, inclusive en aquellos con intervalo QTc normal y test genético positivo. Los de mayor preferencia son nadolol y propranolol, siendo la mayor dosis tolerada por el paciente de acuerdo a su peso, edad y su respuesta individual dada por el conocido espectro de polimorfismo de los receptores beta- adrenérgicos.La indicación de cardiodesfibrilador automático implantable, debe ser como prevención secundaria en pacientes bajo tratamiento farmacológico adecuado que han sufrido de muerte súbita frustra. La prevención primaria en pacientes de alto riesgo siempre debe evaluarse cuidadosamente por el especialista debido a las complicaciones frecuentes de esta terapia en pacientes pediátricos.La simpatectomía lateral izquierda es una terapia segura y eficaz en pacientes pediátricos, que ayuda a disminuir la incidencia de arritmias y puede utilizarse como adyuvante a la terapia con betabloqueantes o ante la intolerancia de los mismos. Otras terapias farmacológicas guiadas de acuerdo a la variante genética se deben considerar con un manejo exclusivo del electrofisiólogo pediátrico.
BIBLIOGRAFIA
- Moss AJ, Schwartz PJ, Crampton RS, et al. The long QT syndrome. Prospective longitudinal study of 328 families. Circulation. 1991; 84:1136–44. [PubMed]
- Schwartz PJ, Stramba- Badiale M, Crotti L, et al. Prevalence of the congenital long-QT syndrome. Circulation 2009;120:1761–1767
- Curran ME, Splawski I, Timothy KW, et al. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 1995; 80:795–803.
- Ackerman MJ, PrioriSG, WillemsS, etal. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association(EHRA).Heart Rhythm 2011;8:1308–1339.
- SchwartzPJ,CrottiL.QTc behavior during exercise and genetic testing for the long-QT syndrome.Circulation2011;124:2181–2184.
- 18. SchwartzPJ, MossAJ, VincentGM, etal. Diagnostic criteria for the long QT syndrome. An update. Circulation1993; 88:782–784.
- Chang IK, Shyu MK, Lee CN, Kau ML, Ko YH, Chow SN, et al. Prenatal diagnosis and treatment of fetal long QT syndrome: a case report. Prenat Diagn. 2002; 22:1209-12.
- Cowan JC, Yusoff K, Moore M, Amos PA, Gold AE, Bourke JP, et al. Importance of lead selection in QT interval measurement. Am J Cardiol. 1988;61:83-7.
- 38. Monnig G, Eckardt L, Wedekind H, Haverkamp W, Gerss J, Milberg P, et al. Electrocardiographic risk stratification in families with congenital long QT syndrome. Eur Heart J. 2006.
- Bazett H. An analysis of the time-relations of electrocardiograms. Heart. 1920;7:353-70.
- Viskin S, Rosovski U, Sands AJ, Chen E, Kistler PM, Kalman JM, et al. Inaccurate electrocardiographic interpretation of long QT: the majority of physicians cannot recognize a long QT when they see one. Heart Rhythm. 2005;2:569-74.
- Beinder E, Grancay T, Menendez T, Singer H, Hofbeck M. Fetal sinus bradycardia and the long QT syndrome. Am J Obstet Gynecol 2001;185:743-7.
- Lupoglazoff JM, Denjoy I, Villain E, Fressart V, Simon F, Bozio A, et al. Long QT syndrome in neonates: conduction disorders associated with HERG mutations and sinus bradycardia with KCNQ1 mutations. J Am Coll Cardiol. 2004;43:826-30.
- Schwartz PJ, Priori SG, Locati EH, Napolitano C, Cantu F, Towbin JA, et al. Long QT syndrome patients with mutations of the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increases in heart rate.Implications for gene-specific therapy. Circulation.1995;92:3381-





































1 Comment
Manuel Felipe
enero 23, 2019, 1:45 pmMuy interesante. Resumido y muy acertado. ¿Sería posible tenerlo en pdf?. Gracias.
REPLY